

Nature:电子催化的分子识别

分子识别和自组装是生命体系结构和功能的基础,对这些过程的深入理解和精准调控,不仅是学习模仿自然界的必由之路,而且提供了“自下而上”创制新材料的有效途径。在化学家看来,催化是最有力的调控手段之一。过往数百年间,各类催化剂层出不穷,不断提高化学反应的效率和选择性,深刻地影响了人类文明。但迄今为止,想要催化分子间的识别和组装,仍然是艰巨的挑战。在为数不多的催化组装实例中,催化剂往往来自于意外发现,而且结构复杂,难以拓展和优化。导致这一现状的主要原因有两点:第一,分子间的作用力强度低,动态性显著,使得分子识别远不如化学反应容易操控;第二,当前自组装领域的研究,对动力学和机理的探讨不够深入,很难为催化剂的理性设计提供指导。
若要发展一类简单、通用的催化组装方法,可以尝试从相对成熟的合成化学领域寻找灵感。近年来,随着自由基反应的复兴,以及光氧化还原催化和有机电合成的蓬勃发展,人们对电子在化学反应中的作用有了更加深刻的认识。2014年,Dennis P. Curran和Armido Studer两位教授联名在Nature Chemistry 上撰文,以“电子是一种催化剂”为题,正式提出了“电子催化”的理念。许多化学反应涉及惰性底物的转化,需要跨越很高的能垒,因而不易发生。如果向体系中注入少量电子,将一部分底物还原成自由基离子,则可以显著地增加反应活性,促进共价键快速的断裂和形成,随之得到的中间体又能够自发地释放出电子,生成产物,完成催化循环。简言之,电子可以充当催化剂的角色,降低反应能垒,加快反应速率。
如果将分子识别看作是一种“非共价反应”,那么遵循相似的机制,电子完全可以用来催化分子间的识别和组装。为了匹配电子催化的原理,组装体系需要满足三个基本条件:第一,具有氧化还原活性,即至少有一种底物能够快速地接受电子;第二,电子注入后可以显著地降低分子识别过程的能垒;第三,存在一种催化中间体,其能够在底物接受电子之后,通过分子间的非共价作用力形成,然后迅速释放电子,转化为组装产物。
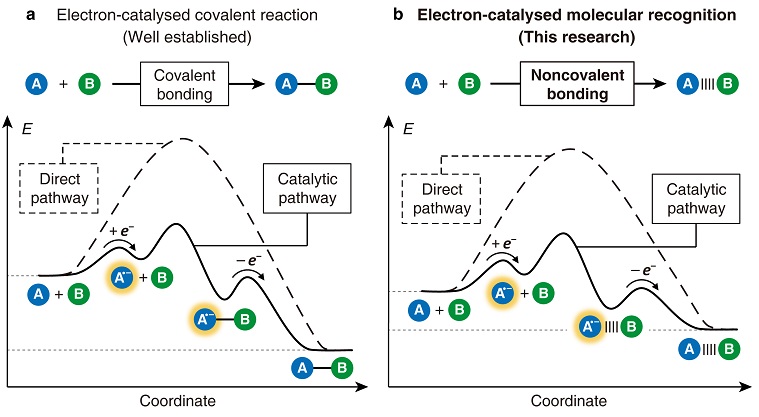
图1. 从电子催化的化学反应到电子催化的分子识别。
近日,美国西北大学的J. Fraser Stoddart教授(2016年诺贝尔化学奖得主)和Michael R. Wasielewski教授、加州理工学院William A. Goddard教授以及缅因大学R. Dean Astumian教授四个团队共同合作,首次实现了电子对分子识别过程的催化和调控。作为一种全新的组装策略,电子催化原理清晰,体系简洁,方法可控,既减轻了对催化剂的结构依赖,又赋予了精准调控组装进程和产物分布的能力。相关论文发表在Nature 上,第一作者为Stoddart课题组的焦阳博士和邱允俨博士。
为了实现电子催化,作者设计了一个自由基状态的主客体系统。其中主体R2(•+)是一种带正电的大环分子,包含两个独立的紫精自由基阳离子(BIPY•+)。客体D+(•+)由三部分构成:中心的BIPY•+单元,可以通过自由基配对作用与R2(•+)结合,形成三自由基复合物(trisradical complex);右侧是一个尺寸很大的空间位阻基团,会完全阻止大环分子的穿过;左侧的吡啶盐基团,则成为分子识别唯一可能的通道。前期探索表明,尽管R2(•+)和D+(•+)之间的结合是热力学有利的,但由于彼此间强烈的静电排斥力,这一过程动力学禁阻,几乎无法进行。作者设想,如果向体系中注入一个电子,R2(•+)和D+(•+)均有可能被还原,其中某一个BIPY•+单元转化为中性的BIPY[0]。由此,体系的电荷数减少,两种分子之间的静电排斥力会明显减弱,从而降低大环分子穿过吡啶盐基团的能垒,迅速形成一种双自由基复合物 [D⊂R]+2(•+)。这一催化中间体能够自发地将电子释放出来,转变为最终的组装产物,即三自由基复合物 [D⊂R]+3(•+),而释放出的电子继续还原新的底物,启动下一个催化循环。根据以上的分子设计和机理假设,作者认为,只需要引入催化量的电子,就可以显著加快两种分子间的识别和组装。
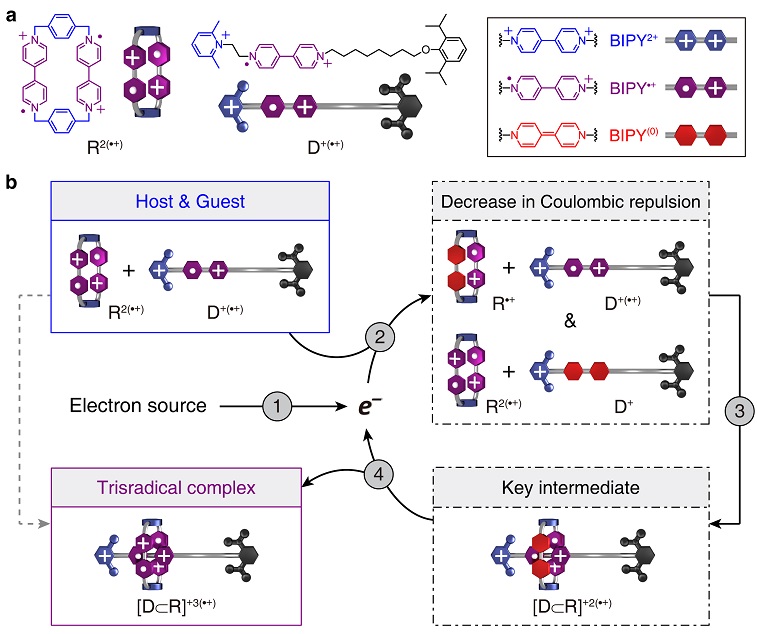
图2. 文中主客体系统的化学结构,以及电子催化的分子识别机理。
为了检验电子催化的效果,作者分别制备了R2(•+)、D+(•+)的乙腈溶液,确保样品中所有的BIPY单元都恰好处在自由基的状态。将R2(•+)和D+(•+)等比例混合,通过UV-Vis-NIR光谱监测,长达10个小时未发现明显变化,说明分子识别几乎没有发生。如果加入4 mol%的CoCp2(二茂钴,一种化学还原剂,能够将BIPY•+还原至BIPY[0]),作者观察到,在1080 nm处出现了三自由基复合物 [D⊂R]+3(•+)的特征吸收峰,该信号随着时间推移不断上升,表明分子识别逐渐进行,并在70分钟内达到平衡。进一步增加CoCp2的用量,分子识别进行得更快,10分钟之内即可完成,速率提高了3200倍。比较动力学曲线可以看出,CoCp2用量越多,分子识别的速率越快,但最终达到的平衡位置基本相同。以上现象充分说明:通过化学还原法注入的电子,能够有效地催化分子识别。
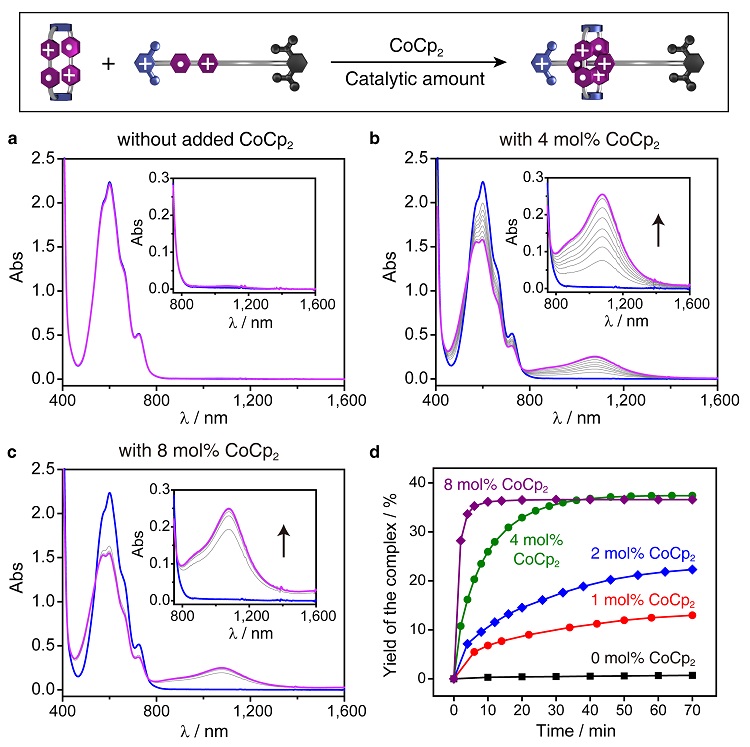
图3. 动力学实验表明,通过化学还原注入的电子可以显著加速分子识别的进行,并且几乎不改变平衡位置。
接下来,作者开展了深入的机理研究。其中一项关键的实验是:将原本自动运转的催化过程,人为拆分成两个步骤。通过这种方式,可以在UV-Vis-NIR光谱中明显观察到催化中间体的形成及其后续转化。进一步,借助模型化合物,证实了该中间体确为双自由基复合物[D⊂R]+2(•+),这一物种具有反磁性,特征的光谱吸收峰位于1700 nm附近,形成的驱动力来源于自由基配对作用和电荷转移相互作用的协同。借助一系列机理实验,辅以量子力学计算,作者清晰地阐释了催化路径,并充分证明了这一催化现象的本质,在于电子的注入有效降低了分子识别过程的能垒。
基于明确的催化机理,作者初步展示了电子催化的一些优势。首先,使用电子作为催化剂,可以不局限于某一种特定的引发剂。理论上,只要具有足够的给电子能力,任何化学试剂都可以引发电子催化。作者筛选了10种还原电位合适的试剂,包括活泼金属、金属配合物和纯有机分子,它们均可以加速分子识别,且效率几乎相同。不同于以往的催化组装体系,电子催化策略能够大幅度缓解对催化剂的结构依赖,使得该方法具有很好的兼容性和实用性。
其次,电子催化可以通过电化学的手段引发和控制。为了说明这一点,作者使用了一种无隔膜的电解池(undivided cell),将阴阳两极置于同一份R2(•+)和D+(•+)的混合溶液中。一旦通电,体系中的BIPY•+单元会同时在阴极接受电子,在阳极释放电子,生成等量的BIPY[0]和BIPY2+。实际效果相当于“空间分离”的歧化反应。这种氧化还原状态的改变是暂时的,当BIPY[0]和BIPY2+远离电极,在溶液中相遇,两者会自发地发生逆歧化反应,重新回到BIPY•+。歧化的速率由电流强度控制,而逆歧化的速率则主要由传质(例如,搅拌引起的对流)控制。因此,在合适的通电条件下,体系达到稳态,可以维持一定浓度的BIPY[0]单元存在,催化R2(•+)和D+(•+)之间的分子识别,生成三自由基复合物[D⊂R]+3(•+)。一旦断电,体系的氧化还原状态完全恢复,换言之,临时注入的电子被移除,催化会立即停止。借助这样的电化学手段,可以使分子识别过程达到“令行禁止”的效果,精确控制速率和转化率,从而获得底物与产物以任意比例分布的一系列动力学亚稳态,这在传统的自组装体系中是很难实现的。
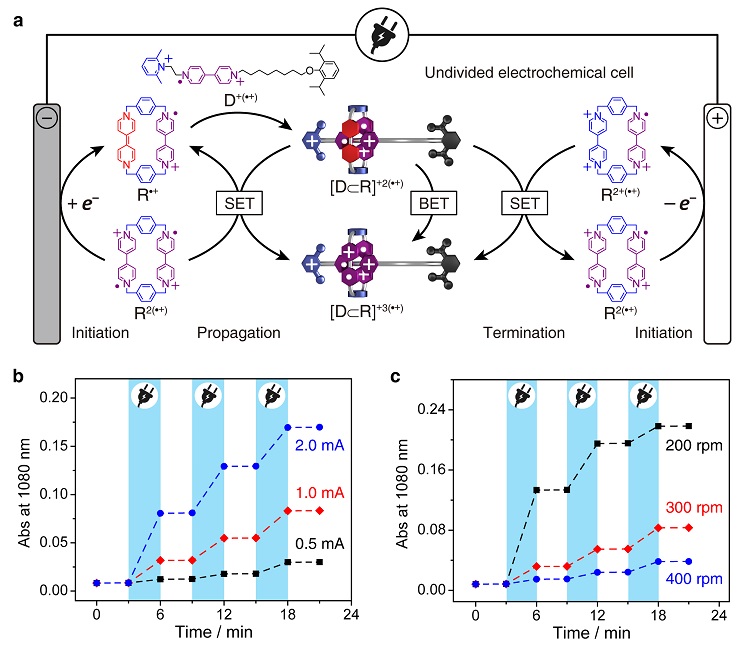
图4. 基于电子催化的原理,运用电化学手段精准控制分子识别的进程。
总之,这项研究将电子催化的理念从合成化学领域拓展到了超分子化学领域,释放出电子在促进和调控自组装方面的巨大潜力。展望未来,这种简洁、高效、通用的催化组装策略,将成为化学家和生物学家手中便捷的工具,用以精准调控各类分子间的识别,引导微观、介观、宏观等多个尺度上的自组装行为,并最终创造出全新的物质和材料。






